
ERN ReCONNET
Disease Info
Disease: IgG4-related disease
IgG4-related disease (IgG4-RDs)
Overview – What is IgG4-RD?
Clinical overview of the disease
IgG4-related disease (IgG4-RD) is characterized by mass-forming lesions with a relapsing-remitting course that might lead to organ failure if left untreated. This fibro-inflammatory disorder is named for the accumulation of IgG4 secreting plasma cells in tissues and the increase of IgG4 concentration in the serum of most patients. First described in 2001 in the setting of autoimmune pancreatitis, IgG4-RD is now known to affect virtually any anatomical district.
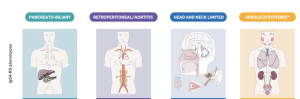
he following phenotypes of IgG4-RD have been identified based on homogeneous epidemiological features, serological findings, and prognostic outcomes:
- Pancreato-hepatobiliary disease
- Retroperitoneal fibrosis, with or without aortitis
- Head and neck limited disease
- Classic Mikulicz’s syndrome with systemic involvement.
These classifications may offer the opportunity to establish personalized monitoring and therapeutic strategies in the near future.
Epidemiology
IgG4-RD predominantly affects middle-aged individuals, with a male/female ratio of approximately 5:1. The incidence and prevalence of IgG4-RD still remain largely underestimated. Based on a recent claims-data analysis from the US, the incidence was 1.39 per 100 000 person-years, and the prevalence 5.3/100 000 persons, respectively in 2019. According to a national epidemiological survey from Japan, the incidence of autoimmune pancreatitis in 2016 was 3.1 cases per 100,000 individuals, but pancreatic involvement represents only one of more than a dozen organs potentially affected by IgG4-RD, underscoring our poor assessment of the epidemiology of the disease in its multiple manifestations.
Risk factors and possible causes
There are no established risk factors associated with the development of IgG4-RD. There are few if any reports, for example, of relatives (of any degree) affected by IgG4-RD indicating that IgG4-RD is not an inherited condition and carries a negligible risk of parental transmission. Similarly, among environmental risk factors, an increased incidence of cigarette smoking and exposure to metals/toxics/poisons (namely, being a «blue collar worker») has been observed in IgG4-RD patients.
The precise mechanisms driving IgG4-RD still remain unclear and are currently being investigated. Aberrant innate and adaptive immunity are considered as the main pathogenesis of IgG4-RD. In general, IgG4-RD follows a biphasic progression with an “inflammatory” phase that culminates in a “fibrotic” outcome. B lymphocytes and plasma cells, immune cells responsible for the production of antibodies, seem to be central to IgG4-RD pathogenesis because B-cell depletion with available immunosuppressive drugs leads to rapid improvement in most cases.
IgG4-related disease (IgG4-RD) is characterized by mass-forming lesions with a relapsing-remitting course that might lead to organ failure if left untreated. This fibro-inflammatory disorder is named for the accumulation of IgG4 secreting plasma cells in tissues and the increase of IgG4 concentration in the serum of most patients. First described in 2001 in the setting of autoimmune pancreatitis, IgG4-RD is now known to affect virtually any anatomical district.
he following phenotypes of IgG4-RD have been identified based on homogeneous epidemiological features, serological findings, and prognostic outcomes:
IgG4-RD predominantly affects middle-aged individuals, with a male/female ratio of approximately 5:1. The incidence and prevalence of IgG4-RD still remain largely underestimated. Based on a recent claims-data analysis from the US, the incidence was 1.39 per 100 000 person-years, and the prevalence 5.3/100 000 persons, respectively in 2019. According to a national epidemiological survey from Japan, the incidence of autoimmune pancreatitis in 2016 was 3.1 cases per 100,000 individuals, but pancreatic involvement represents only one of more than a dozen organs potentially affected by IgG4-RD, underscoring our poor assessment of the epidemiology of the disease in its multiple manifestations.
There are no established risk factors associated with the development of IgG4-RD. There are few if any reports, for example, of relatives (of any degree) affected by IgG4-RD indicating that IgG4-RD is not an inherited condition and carries a negligible risk of parental transmission. Similarly, among environmental risk factors, an increased incidence of cigarette smoking and exposure to metals/toxics/poisons (namely, being a «blue collar worker») has been observed in IgG4-RD patients.
The precise mechanisms driving IgG4-RD still remain unclear and are currently being investigated. Aberrant innate and adaptive immunity are considered as the main pathogenesis of IgG4-RD. In general, IgG4-RD follows a biphasic progression with an “inflammatory” phase that culminates in a “fibrotic” outcome. B lymphocytes and plasma cells, immune cells responsible for the production of antibodies, seem to be central to IgG4-RD pathogenesis because B-cell depletion with available immunosuppressive drugs leads to rapid improvement in most cases.
Emanuel Della Torre
Emanuel Della Torre
Disease Coordinator
Disease Coordinator
Common symptoms
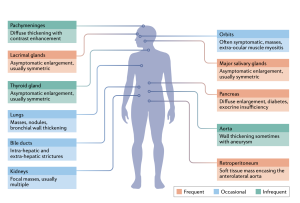
The clinical presentation of IgG4-RD is usually indolent, with signs and symptoms becoming evident over months or even years. Most characteristic are pseudotumor-like lesions involving single or multiple organs. Different organs might be affected at the same time or one after the other. Clinical manifestations are non-specific, vary according to the organs, and relate to the mechanical compression exerted by the fibrotic masses on local structures.
The most common manifestations include autoimmune pancreatitis, chronic periaortitis, retroperitoneal fibrosis and salivary or lachrymal gland swelling. Atypical presentations of IgG4-RD include tubulointerstitial nephritis, glomerulonephritis, interstitial lung disease, pleural and pericardial effusion.
Diagnosis (path/ tests / differential diagnosis)
Definitive diagnosis of IgG4-RD requires rigorous clinical-pathological correlation because clinical assessments, laboratory evaluations, and imaging studies are often insufficient to distinguish neoplastic, inflammatory and infectious mimickers.
Serological findings in patients with IgG4-RD are largely non-specific. Serum IgG4 elevation occurs in 55 to 97% of cases and correlates with the number of organs involved and the risk of relapse. However, an increase in serum IgG4 has poor specificity for diagnostic purposes because it can be observed in a broad spectrum of neoplastic, infectious, and autoimmune diseases.
Radiological findings are usually nonspecific in most affected organs except in case of IgG4-related pancreatitis where imaging classically shows a diffusely enlarged ‘sausage-shaped’ pancreas with a surrounding halo of edematous tissue. Because of the shortcomings of serological and radiological findings, histological examination remains the mainstay for definitive diagnosis and should be performed whenever possible. Characteristic findings comprise dense lymphoplasmacytic infiltration together with tissue fibrosis, and obliterative phlebitis.
Immunostaining is an essential part of the histological diagnosis of IgG4-RD, requiring a careful assessment in three aspects: the number, ratio, and distribution of IgG4+ plasma cells. To overcome these diagnostic challenges, experts developed the 2019 “ACR/EULAR Classification Criteria for IgG4-Related Disease” whereby a patient is classified if a cumulative score of ≥ 20 points is obtained.
 Treatments overview
Treatments overview
In general, treatment is not required for all patients with IgG4-RD, and watchful waiting is prudent in some cases.
When vital organs are involved or patients become symptomatic, treatment is needed to prevent organ dysfunction and failure. Response to treatment might be assessed by means of the IgG4-RD Responder Index, a validated tool for monitoring clinical, radiological and serological outcomes of IgG4-RD.
Glucocorticoids represent the first-line therapy because they lead to dramatic clinical responses in the majority of cases.
A variety of steroid-sparing agents have been employed in different anatomical districts as remission-maintenance drugs (e.g. azathioprine, mycophenolate mofetil, methotrexate, cyclophosphamide).
In addition, the anti-CD20 monoclonal antibody rituximab has been proved to induce swift clinical responses and a decline of serum IgG4 titre in patients with recurrent or refractory disease. When urgent decompression is needed, as in the case of biliary duct or ureteral strictures, temporary stenting or surgical approaches remain the strategy of choice for preventing organ damage
Prognosis
If left untreated IgG4-RD can lead to severe organ dysfunction.
Long-term prognosis largely depends on the affected organs and on a timely institution of adequate immunosuppressive therapy in order to prevent fibrotic damage. IgG4-RD has a marked tendency to relapse, especially in patients with elevated serum IgG4 at baseline, multiorgan involvement, and a history of disease relapse.
When to see a doctor?
IgG4-RD can be suspected in case of one or more gland or organ swelling, especially in case of salivary/lacrimal gland or pancreatic enlargement that might be clinically evident or identified on imaging studies. Serum IgG4 elevation may increase the probability of having IgG4-RD but histological examination if required to rule important differential diagnoses such as malignancies. “Red flags” that may trigger the suspicion for the disease are currently investigated.
Lifestyle tips
– Avoid being overweight:
- Have a balanced and healthy diet – for more info, please click here
- Consume less salt, saturated fats and sugar
- Exercise regularly – at least 150 minutes a week of moderate-intensity physical activity (30 minutes per day)
– Quit smoking
– Reduce alcohol consumption
– Get vaccinated
– Stay engaged with your community
– Have a work and family life balance
– Do something you feel passionate about


Written by Geoff Case, digital editor, RARE Revolution Magazine. Interview with Ilaria Galetti, patient advocate
ERN ReCONNET publications are available for consultation.
LIST OF USEFUL REFERENCES
- Identification of red flags for IgG4-related disease: an international European Reference Network for Rare Connective Tissue Diseases framework
Della-Torre E, Talarico R, Ballarin J
Lancet Rheumatol. 2025 Jan;7(1):e64-e71. doi: 10.1016/S2665-9913(24)00192-9. Epub 2024 Oct 29. PMID: 39486422.
- Inebilizumab for Treatment of IgG4-Related Disease.
Stone JH, Khosroshahi A, Zhang W, Della Torre E, Okazaki K, Tanaka Y, Löhr JM, Schleinitz N, Dong L, Umehara H, Lanzillotta M, Wallace ZS, Ebbo M, Webster GJ, Martinez Valle F, Nayar MK, Perugino CA, Rebours V, Dong X, Wu Y, Li Q, Rampal N, Cimbora D, Culver EL; MITIGATE Trial Investigators. N Engl J Med. 2025 Mar 27;392(12):1168-1177.
- Advances in the diagnosis and management of IgG4 related disease.
Lanzillotta M, Mancuso G, Della-Torre E.
BMJ. 2020 Jun 16;369:m1067. doi: 10.1136/bmj.m1067. PMID: 32546500.
- IgG4-related disease: an update on pathophysiology and implications for clinical care
Perugino C.A. & Stone J.H.
Nature Reviews Rheumatology volume 16, pages702–714 (2020)
- International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease
Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al.
Arthritis Rheumatol. 2015 Jul;67(7): 1688-99.
- Consensus statement on the pathology of IgG4-related disease
Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al.
Mod Pathol. 2012 Sep;25(9):1181-92.
- Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations
Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, Stone JH, al.
Arthritis Rheum. 2012 Oct;64(10):3061-7.
Della-Torre E, Talarico R, Ballarin J
Lancet Rheumatol. 2025 Jan;7(1):e64-e71. doi: 10.1016/S2665-9913(24)00192-9. Epub 2024 Oct 29. PMID: 39486422.
Stone JH, Khosroshahi A, Zhang W, Della Torre E, Okazaki K, Tanaka Y, Löhr JM, Schleinitz N, Dong L, Umehara H, Lanzillotta M, Wallace ZS, Ebbo M, Webster GJ, Martinez Valle F, Nayar MK, Perugino CA, Rebours V, Dong X, Wu Y, Li Q, Rampal N, Cimbora D, Culver EL; MITIGATE Trial Investigators. N Engl J Med. 2025 Mar 27;392(12):1168-1177.
Lanzillotta M, Mancuso G, Della-Torre E.
BMJ. 2020 Jun 16;369:m1067. doi: 10.1136/bmj.m1067. PMID: 32546500.
Perugino C.A. & Stone J.H.
Nature Reviews Rheumatology volume 16, pages702–714 (2020)
Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al.
Arthritis Rheumatol. 2015 Jul;67(7): 1688-99.
Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al.
Mod Pathol. 2012 Sep;25(9):1181-92.
Stone JH, Khosroshahi A, Deshpande V, Chan JK, Heathcote JG, Aalberse R, Stone JH, al.
Arthritis Rheum. 2012 Oct;64(10):3061-7.
IgG4 centres in ERN ReCONNET
Helsinki University Hospital, Hospital District of Helsinki and Uusimaa, Finland (ReCONNETFIN) (adult and paedriatric)
