
ERN ReCONNET
Disease Info
Disease:Idiopathic Inflammatory Myopathies
Idiopathic Inflammatory Myopathies (IIMs)
Overview – What are IIMs?
Clinical overview of the diseases
Idiopathic inflammatory myopathies (IIMs) are a large heterogeneous group of autoimmune diseases whose common denominator is the involvement of striated skeletal muscles.
IIMs are chronic, systemic diseases that may also affect the skin, joints and lungs. Recent advances have led to a subdivision of this group into four main categories:
- Dermatomyositis
- Immune-mediated necrotizing myopathies
- Inclusion body myositis
- Overlap myositis, including antisynthetase syndrome and scleromyositis
Epidemiology
IIMs incidence is about 8 cases per million inhabitants per year. They have been described in the five continents. It has increased during the last 50 years, which may reflect progress in diagnostic performance but also true modification of the epidemiology. Adult onset is about 10-fold more frequent than juvenile onset. Women are about 2-fold more frequently affected than men.
Clinical overview of the disease
Myopathy (proximal muscle weakness, dysphagia and/or elevated serum levels of creatine kinase) is the common denominator of IIMs. However, IIMs are systemic disease whose symptoms also include skin rash, polyarthritis, interstitial lung disease, Raynaud’s phenomenon and/or myocarditis. These extra-muscular symptoms can inaugurate the disease while myopathy can be absent (amopathic forms) or may be very mild (hypomyopathic forms).
Risk factors and possible causes
The causes of IIMs are partially known and depend on the subtypes of IIMs.
Immunogenetics factors explain only about 20% of the disease. IIMs are not genetic myopathies.
Ingrid Lundberg
Ingrid Lundberg
Disease Coordinator
Disease Coordinator

Congrats to our new ePAGs Magdalena, Camelia, Olga, and Silke. Welcome on board!
Alain Meyer
Alain Meyer
Disease Coordinator
Disease Coordinator

Several environmental factors have been highlights and linked with different subgroups of IIMs:
- A cancer is associated with IIMs in 10% of the case (especially dermatomyositis and necrotizing myopathy without antibodies, see below “diagnosis”)
- Ultraviolet radiation plays a role in the onset and relapses of dermatomyositis
- Viral infections have been linked with the onset of several subtypes of IIMs
- Some drugs have been associated with the onset of IIMs. The most common are statins (associated with autoimmune necrotizing myopathies with anti-HMGCR autoantibodies) and immune checkpoint inhibitors (associated with a peculiar form of IIMs)
- Smoking has been involved in antisynthetase syndrome with anti-Jo1 antibodies
 MULTIDISCIPLINARY DIAGNOSIS AND MULTIDISCIPLINARY TEAMS TREATMENT IN IDIOPATHIC INFLAMMATORY MYOPATHIES (IIM)
MULTIDISCIPLINARY DIAGNOSIS AND MULTIDISCIPLINARY TEAMS TREATMENT IN IDIOPATHIC INFLAMMATORY MYOPATHIES (IIM)
Speakers Prof. Louise Diederichsen, ePAGs Olga Drápalová and Silke Schlüter, moderator Prof. Ingrid Lundberg.
Sept. 25th, 2024 – 16.00 CEST.
Common symptoms
– Myopathy in IIMs typically affects the upper and lower limb–girdles. It consists in weakness, leading to difficulty to walk, climb stairs and carry heavy loads. In up to two third of the cases, myopathy affects swallowing muscles (dysphagia). The severity of the myopathy varies widely, ranging from normal strength to loss of walking ability and/or impossibility to eat. Myalgia can be present or absent, being not a reliable sign of myopathy.
The serum levels of creatine kinase (a muscle enzyme) is generally increased. The onset is generally over several months. Yet, in some cases, myopathy occurs over several years that can lead to a misdiagnosis of a genetic myopathy. A peculiar form of IIMs, inclusion body myositis, is characterized by weakness prevailing on finger flexors and quadriceps muscles.
– Skin involvement is present in half of the patients. It consists of a rash over the eyelids (heliotrope rash), rashes and/or papules on the extensor surfaces of joints (Gottron’s sign and/or papules) that are mostly seen in patients with dermatomyositis. It can masquerade lupus rash. Hyperkeratosis and fissures of the fingers of the hands (mechanic’s hands) and/or the feet (hiker’s feet) are mainly seen in antisynthetase syndrome. It can masquerade psoriasis. Skin thickening is mainly seen in scleromyositis.
– Joint involvement affects 30% of the patients. It consists of inflammatory arthralgia and/or arthritis of the small joints of the hands and/or feet. It is more frequent in antisynthetase syndrome, dermatomyositis and scleromyositis. It can masquerade rheumatoid arthritis, but is usually non-erosive.
– Lung involvement affects 30% of the patients. It consists of cough and/or dyspnea. It is more frequent in antisynthetase syndrome, dermatomyositis and scleromyositis. A high resolution chest CT scan and pulmonary function tests are necessary to diagnose and monitor this involvement. It can masquerade idiopathic fibrosis
– Raynaud’s phenomenon affects 30% of the patients. It is more frequent in antisynthetase syndrome, dermatomyositis and scleromyositis. Digital ulcer tips can occur. It can be distinguished from primary Raynaud’s phenomenon with capillaroscopy.
– Heart involvement affects about 9 % of the patients. It consists of chest pain, dyspnea and or stroke. Echocardiography, ECG and sometimes cardiac MRI are needed to diagnose and monitor this involvement.
When to see a doctor
Weakness, skin rash, arthralgia, cough, dyspnea, Raynaud’s phenomenon are the main signs that should lead to a consultation.
Diagnosis (PATH/ TESTS / DIFFERENTIAL DIAGNOSIS)
In a patient with one or more of the signs listed above, two diagnostic tools should be considered. They should be performed in an expert center for IIMs.
Autoantibodies detection. IIMs are autoimmune diseases. The majority of the patients have autoantibodies that can be detected in the blood. About twenty different autoantibodies can be detected with commercial kits and have been shown to be useful for diagnosing and classifying IIMs.
Recorded Webinars
The most frequent autoantibodies in IIMs are:
- Dermatomyositis: anti-Mi2, -TIF1-y, -NXP2, -SAE, -MDA5
- Antisynthetase syndrome: anti-Jo-1, -PL7, -PL12, -OJ, -EJ, -KS, -Ha, -Zo
- Autoimmune necrotizing myopathies: anti-SRP, -HMGCR
- Scleromyositis: anti-U1-RNP, -PM/Scl, -Ku, -U3-RNP
In around 20% of patients, no autoantibodies are detected. These patients are believed to have autoantibodies which have not yet been discovered or for which no available detection technique exists in current practice.
Muscle biopsy. Muscle biopsy consists in the removal of a small piece of the skeletal muscle for histological (and sometimes biochemical and/or molecular examination). Despite the progress in autoantibodies detection, it is still a crucial step in the diagnosis and the classification of IIMs.
Treatment overview
Treatment should be started as soon as possible in coordination with an expert center for IIMs. The aims of the treatment are to:
– Induce and maintain remission (or low disease activity) of the disease. The control of the activity of the disease is mandatory in order to restore functionality and to prevent disease damages (see below). This aim will be achieved using:
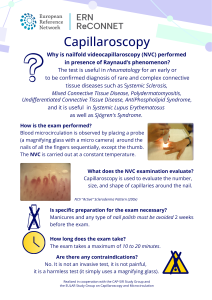 The ERN ReCONNET Education and Training Working Group (WG) is glad to release its new flyer on nailfold videocapillaroscopy (NVC), a new and informative document targeting patients and lay audience aimed at explaining such an important diagnostic approach.
The ERN ReCONNET Education and Training Working Group (WG) is glad to release its new flyer on nailfold videocapillaroscopy (NVC), a new and informative document targeting patients and lay audience aimed at explaining such an important diagnostic approach.
Regarding the NVC approach, the test is mainly useful in rheumatology and in internal medicine, for an early or to-be-confirmed diagnosis of rare and complex connective tissue diseases such as System Sclerosis (SSc), Mixed Connective Tissue Disease (MCTD), Polydermatomyositis, Undifferentiated Connective Tissue Disease (UCTD), AntiPhospholipid Syndrome (APS), and offers important information on microcirculation in Systemic Lupus Erythematosus (SLE) as well as in Sjögren’s Syndrome (SS) and overlaps.
The flyer can be downloaded for free (pdf), and it will soon available in several languages:
ERN ReCONNET Capillaroscopy Flyer (pdf)
More details and info about NVC can be found here.
1-Induction treatment. Corticosteroids are usually used to achieve remission (or low disease activity). They are generally given at high dose (1 mg/kg/day orally, sometimes in intravenous bolus). To limit the risk of long-term side effects of corticosteroids, these drugs are gradually tapered and eventually discontinued when possible.
2-Maintenance treatment. To maintain remission upon corticosteroids tapering, immunomodulatory drugs are prescribed, generally starting at the same time as corticosteroids. These drugs will be pursued for several years. The choice of the immunomodulatory drug depends on the subtype of the IIMs and of the organ involvement. The prevention of the flares with photo-protection (especially in dermatomyositis), tobacco cessation (especially in antisynthetase syndrome) are also important measures.
– Limit and reverse the damage of the disease. As opposed to disease activity, damage is the cumulation of residual manifestations associated with sequelae that do not respond to immunomodulators. They include sarcopenia, skin depigmentation, calcinosis… Early initiation and tight control of the disease limits the risk of damage. Exercise is effective to reverse muscle damage and should be systematically implemented. Surgery can be used to treat calcinosis.
– Research and treat an associated cancer. In 10% of the patients, a cancer is associated with IIMs. In these cases, the treatment of the cancer will generally also treat the IIM. The risk of an associated cancer depends on the IIMs subtype. Cancer screening and monitoring should be performed at diagnosis and in some cases up to 3 years after diagnosis depending on the risk.
– Restore functionality. Myositis can have a dramatic impact on functionality and activities of daily life. Early treatment, including active physical and occupational therapy from the start can minimize this impact.
– Prevent comorbidities. The risk of infections, cardiovascular events and/or osteoporosis is increased in IIMs because of the disease and its treatments. This risk can be decreased with vaccinations, diet, exercises and some preventive drugs when needed.
– Cope with the disease. Therapeutic education, and interaction with peers through patients’ associations are crucial supports to cope with IIMs.
Prognosis
Current treatments are generally needed for several years to maintain the control of IIMs. Several studies have indicated that the prognosis of IIMs has improved over the last decades. Yet, important improvements in the delay of diagnosis and the current conventional cares are needed to increase life expectancy and quality of life that are still significantly affected by these diseases.
Patient organisations

Denmark. The myositis group in under the umbrella of the Arthritis Association
Tove Wolf – tovewolf(at)mail.tele.dk – Linda Rath – lindarath(at)email.dk
Czech Republic. The Czech Myositis Working Goup (CMWK) operates under the umbrella of the Czech League against Rheumatism (CLAR)
Olga Drápalová – olga.drapalova(at)revmaliga.cz
Germany. The German Myositis-Group (Myositis-Gruppe) within the German Society for Neuromuscular Diseases (Deutsche Gesellschaft für Muskelkranke e.V. – DG)
Silke Schlüter: silke.schlueter(at)dgm.org
Dutch. Dutch Myositis Working Group (DMWG) under the umbrella of Spierziekten Nederland (Dutch Patient association for neuromuscular diseases)
Ingrid de Groot: Ingrid.de.groot(at)upcmail.nl
Sweden. The Swedish Myositis Working Group (Riksförening för myositsjukdomar) within the Swedish Rheumatism Association (Reumatikerförbundet)
Anneli Dihkan – anneli(at)eneby.nu
United Kingdom. Myositis UK
Irene Oakley – irene(at)myositis.org.uk
Begium, Flemish. CIB-Liga (Chronische Inflammatoire Bindweefselziekten), under the umbrella of ReumaNet
Portugal. myositis group is under umbrela of – Associação Portuguese de Doentes Neuromusculares
France. AFM TELETHON – Groupe d’intérêt GIMI
Anne-Elisabeth LAUNAY – aelaunay(at)afm-telethon.fr
Additional ERN ReCONNET publications are available for consultation.
LIST OF USEFUL REFERENCES
1. International Guideline for Idiopathic Inflammatory Myopathy-Associated Cancer Screening: an International Myositis Assessment and Clinical Studies Group (IMACS) initiative.
Oldroyd AGS, Callen JP, Chinoy H, Chung L, Fiorentino D, Gordon P, Machado PM, McHugh N, Selva-O’Callaghan A, Schmidt J, Tansley SL, Vleugels RA, Werth VP; International Myositis Assessment and Clinical Studies Group Cancer Screening Expert Group; Aggarwal R.
Nat Rev Rheumatol. 2023 Dec;19(12):805-817.
2. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) Guideline for the Screening and Monitoring of Interstitial Lung Disease in People with Systemic Autoimmune Rheumatic Diseases.
Johnson SR, Bernstein EJ, Bolster MB, Chung JH, Danoff SK, George MD, Khanna D, Guyatt G, Mirza RD, Aggarwal R, Allen A Jr, Assassi S, Buckley L, Chami HA, Corwin DS, Dellaripa PF, Domsic RT, Doyle TJ, Falardeau CM, Frech TM, Gibbons FK, Hinchcliff M, Johnson C, Kanne JP, Kim JS, Lim SY, Matson S, McMahan ZH, Merck SJ, Nesbitt K, Scholand MB, Shapiro L, Sharkey CD, Summer R, Varga J, Warrier A, Agarwal SK, Antin-Ozerkis D, Bemiss B, Chowdhary V, Dematte D’Amico JE, Hallowell R, Hinze AM, Injean PA, Jiwrajka N, Joerns EK, Lee JS, Makol A, McDermott GC, Natalini JG, Oldham JM, Saygin D, Lakin KS, Singh N, Solomon JJ, Sparks JA, Turgunbaev M, Vaseer S, Turner A, Uhl S, Ivlev I.
Arthritis Rheumatol. 2024 Aug;76(8):1201-1213.
3. 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) Guideline for the Treatment of Interstitial Lung Disease in People with Systemic Autoimmune Rheumatic Diseases.
Johnson SR, Bernstein EJ, Bolster MB, Chung JH, Danoff SK, George MD, Khanna D, Guyatt G, Mirza RD, Aggarwal R, Allen A Jr, Assassi S, Buckley L, Chami HA, Corwin DS, Dellaripa PF, Domsic RT, Doyle TJ, Falardeau CM, Frech TM, Gibbons FK, Hinchcliff M, Johnson C, Kanne JP, Kim JS, Lim SY, Matson S, McMahan ZH, Merck SJ, Nesbitt K, Scholand MB, Shapiro L, Sharkey CD, Summer R, Varga J, Warrier A, Agarwal SK, Antin-Ozerkis D, Bemiss B, Chowdhary V, Dematte D’Amico JE, Hallowell R, Hinze AM, Injean PA, Jiwrajka N, Joerns EK, Lee JS, Makol A, McDermott GC, Natalini JG, Oldham JM, Saygin D, Lakin KS, Singh N, Solomon JJ, Sparks JA, Turgunbaev M, Vaseer S, Turner A, Uhl S, Ivlev I.
Arthritis Care Res (Hoboken). 2024 Aug;76(8):1051-1069.
4. ERS/EULAR clinical practice guidelines for connective tissue disease-associated interstitial lung disease developed by the task force for connective tissue disease-associated interstitial lung disease of the European Respiratory Society (ERS) and the European Alliance of Associations for Rheumatology (EULAR) Endorsed by the European Reference Network on rare respiratory diseases (ERN-LUNG).
Antoniou KM, Distler O, Gheorghiu AM, Moor CC, Vikse J, Bizymi N, Galetti I, Brown G, Bargagli E, Allanore Y, Corte TJ, Dieudé P, Cottin V, Fisher BA, Fabre A, Giles JT, Kreuter M, Lundberg IE, Poletti V, Maurer B, Renzoni EA, Müller-Ladner U, Strek ME, Sverzellati N, Studenic P, Mohammed J, Nagavci B, Stamm T, Tonia T, Crestani B, Hoffmann-Vold AM.
Ann Rheum Dis. 2026 Jan;85(1):22-60.
5. 2024 ACC Expert Consensus Decision Pathway on Strategies and Criteria for the Diagnosis and Management of Myocarditis: A Report of the American College of Cardiology Solution Set Oversight Committee.
Writing Committee; Drazner MH, Bozkurt B, Cooper LT, Aggarwal NR, Basso C, Bhave NM, Caforio ALP, Ferreira VM, Heidecker B, Kontorovich AR, Martín P, Roth GA, Van Eyk JE.
J Am Coll Cardiol. 2025 Feb 4;85(4):391-431. doi: 10.1016/j.jacc.2024.10.080. Epub 2024 Dec 10. PMID: 39665703.
6. British Society for Rheumatology Standards, Audit and Guidelines Working Group. British Society for Rheumatology guideline on management of paediatric, adolescent and adult patients with idiopathic inflammatory myopathy.
Oldroyd AGS, Lilleker JB, Amin T, Aragon O, Bechman K, Cuthbert V, Galloway J, Gordon P, Gregory WJ, Gunawardena H, Hanna MG, Isenberg D, Jackman J, Kiely PDW, Livermore P, Machado PM, Maillard S, McHugh N, Murphy R, Pilkington C, Prabu A, Rushe P, Spinty S, Swan J, Tahir H, Tansley SL, Truepenny P, Truepenny Y, Warrier K, Yates M, Papadopoulou C, Martin N, McCann L, Chinoy H;
Rheumatology (Oxford). 2022 May 5;61(5):1760-1768.
7. Treatment consensus for management of polymyositis and dermatomyositis among rheumatologists, neurologists and dermatologists.
Kohsaka H, Mimori T, Kanda T, Shimizu J, Sunada Y, Fujimoto M, Kawaguchi Y, Jinnin M, Muro Y, Ishihara S, Tomimitsu H, Ohta A, Sumida T.
Mod Rheumatol. 2019 Jan;29(1):1-19.
8. The Brazilian Society of Rheumatology recommendations on investigation and diagnosis of systemic autoimmune myopathies.
de Souza FHC, de Araújo DB, Vilela VS, Simões RS, Bernardo WM, Frank TA, da Cunha BM, Shinjo SK.
Adv Rheumatol. 2019 Oct 10;59(1):42.
9. Guidelines on dermatomyositis–excerpt from the interdisciplinary S2k guidelines on myositis syndromes by the German Society of Neurology.
Sunderkötter C, Nast A, Worm M, Dengler R, Dörner T, Ganter H, Hohlfeld R, Melms A, Melzer N, Rösler K, Schmidt J, Sinnreich M, Walter MC, Wanschitz J, Wiendl H.
J Dtsch Dermatol Ges. 2016 Mar;14(3):321-38.
10. The Classification Criteria for Anti-Synthetase Syndrome (Class) Project
Zanframundo et al. Arthritis Rheumatol. 2024; 76 (suppl 9).
11. 272nd ENMC international workshop: 10 Years of progress – revision of the ENMC 2013 diagnostic criteria for inclusion body myositis and clinical trial readiness. 16-18 June 2023, Hoofddorp, The Netherlands.
Lilleker JB, Naddaf E, Saris CGJ, Schmidt J, de Visser M, Weihl CC; 272nd ENMC workshop participants.
Neuromuscul Disord. 2024 Apr;37:36-51. doi: 10.1016/j.nmd.2024.03.001.
12. 273rd ENMC International workshop: Clinico-Sero-morphological classification of the Antisynthetase syndrome. Amsterdam, The Netherlands, 27-29 October 2023.
Stenzel W, Mammen AL, Gallay L, Holzer MT, Kleefeld F, Benveniste O, Allenbach Y; ENMC Antisynthetase Syndrome Study Group.
Neuromuscul Disord. 2024 Dec;45:104453.
13. 255th ENMC workshop: Muscle imaging in idiopathic inflammatory myopathies. 15th January, 16th January and 22nd January 2021 – virtual meeting and hybrid meeting on 9th and 19th September 2022 in Hoofddorp, The Netherlands.
de Visser M, Carlier P, Vencovský J, Kubínová K, Preusse C; ENMC Muscle Imaging in Idiopathic Inflammatory Myopathies workshop study group.
Neuromuscul Disord. 2023 Oct;33(10):800-816.
14. 256(th) ENMC international workshop: Myositis specific and associated autoantibodies (MSA-ab): Amsterdam, The Netherlands, 8-10 October 2021.
Damoiseaux J, Mammen AL, Piette Y, Benveniste O, Allenbach Y; ENMC 256th Workshop Study Group.
Neuromuscul Disord. 2022 Jul;32(7):594-608
15. 239th ENMC International Workshop: Classification of dermatomyositis, Amsterdam, the Netherlands, 14-16 December 2018.
Mammen AL, Allenbach Y, Stenzel W, Benveniste O; ENMC 239th Workshop Study Group.
Neuromuscul Disord. 2020 Jan;30(1):70-92
16. 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016.
Allenbach Y, Mammen AL, Benveniste O, Stenzel W; Immune-Mediated Necrotizing Myopathies Working Group.
17. 2016 American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in adult dermatomyositis and polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative
Aggarwal R, Rider LG, Ruperto N, Bayat N, Erman B, Feldman BM, et al.
Ann Rheum Dis. 2017 May;76(5):792-801.
18. Consensus-based recommendations for the management of juvenile dermatomyositis
Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al.
Ann Rheum Dis. 2017 Feb;76(2):329-340.
Oldroyd AGS, Callen JP, Chinoy H, Chung L, Fiorentino D, Gordon P, Machado PM, McHugh N, Selva-O’Callaghan A, Schmidt J, Tansley SL, Vleugels RA, Werth VP; International Myositis Assessment and Clinical Studies Group Cancer Screening Expert Group; Aggarwal R.
Nat Rev Rheumatol. 2023 Dec;19(12):805-817.
Johnson SR, Bernstein EJ, Bolster MB, Chung JH, Danoff SK, George MD, Khanna D, Guyatt G, Mirza RD, Aggarwal R, Allen A Jr, Assassi S, Buckley L, Chami HA, Corwin DS, Dellaripa PF, Domsic RT, Doyle TJ, Falardeau CM, Frech TM, Gibbons FK, Hinchcliff M, Johnson C, Kanne JP, Kim JS, Lim SY, Matson S, McMahan ZH, Merck SJ, Nesbitt K, Scholand MB, Shapiro L, Sharkey CD, Summer R, Varga J, Warrier A, Agarwal SK, Antin-Ozerkis D, Bemiss B, Chowdhary V, Dematte D’Amico JE, Hallowell R, Hinze AM, Injean PA, Jiwrajka N, Joerns EK, Lee JS, Makol A, McDermott GC, Natalini JG, Oldham JM, Saygin D, Lakin KS, Singh N, Solomon JJ, Sparks JA, Turgunbaev M, Vaseer S, Turner A, Uhl S, Ivlev I.
Arthritis Rheumatol. 2024 Aug;76(8):1201-1213.
Johnson SR, Bernstein EJ, Bolster MB, Chung JH, Danoff SK, George MD, Khanna D, Guyatt G, Mirza RD, Aggarwal R, Allen A Jr, Assassi S, Buckley L, Chami HA, Corwin DS, Dellaripa PF, Domsic RT, Doyle TJ, Falardeau CM, Frech TM, Gibbons FK, Hinchcliff M, Johnson C, Kanne JP, Kim JS, Lim SY, Matson S, McMahan ZH, Merck SJ, Nesbitt K, Scholand MB, Shapiro L, Sharkey CD, Summer R, Varga J, Warrier A, Agarwal SK, Antin-Ozerkis D, Bemiss B, Chowdhary V, Dematte D’Amico JE, Hallowell R, Hinze AM, Injean PA, Jiwrajka N, Joerns EK, Lee JS, Makol A, McDermott GC, Natalini JG, Oldham JM, Saygin D, Lakin KS, Singh N, Solomon JJ, Sparks JA, Turgunbaev M, Vaseer S, Turner A, Uhl S, Ivlev I.
Arthritis Care Res (Hoboken). 2024 Aug;76(8):1051-1069.
Antoniou KM, Distler O, Gheorghiu AM, Moor CC, Vikse J, Bizymi N, Galetti I, Brown G, Bargagli E, Allanore Y, Corte TJ, Dieudé P, Cottin V, Fisher BA, Fabre A, Giles JT, Kreuter M, Lundberg IE, Poletti V, Maurer B, Renzoni EA, Müller-Ladner U, Strek ME, Sverzellati N, Studenic P, Mohammed J, Nagavci B, Stamm T, Tonia T, Crestani B, Hoffmann-Vold AM.
Ann Rheum Dis. 2026 Jan;85(1):22-60.
Writing Committee; Drazner MH, Bozkurt B, Cooper LT, Aggarwal NR, Basso C, Bhave NM, Caforio ALP, Ferreira VM, Heidecker B, Kontorovich AR, Martín P, Roth GA, Van Eyk JE.
J Am Coll Cardiol. 2025 Feb 4;85(4):391-431. doi: 10.1016/j.jacc.2024.10.080. Epub 2024 Dec 10. PMID: 39665703.
Oldroyd AGS, Lilleker JB, Amin T, Aragon O, Bechman K, Cuthbert V, Galloway J, Gordon P, Gregory WJ, Gunawardena H, Hanna MG, Isenberg D, Jackman J, Kiely PDW, Livermore P, Machado PM, Maillard S, McHugh N, Murphy R, Pilkington C, Prabu A, Rushe P, Spinty S, Swan J, Tahir H, Tansley SL, Truepenny P, Truepenny Y, Warrier K, Yates M, Papadopoulou C, Martin N, McCann L, Chinoy H;
Rheumatology (Oxford). 2022 May 5;61(5):1760-1768.
Kohsaka H, Mimori T, Kanda T, Shimizu J, Sunada Y, Fujimoto M, Kawaguchi Y, Jinnin M, Muro Y, Ishihara S, Tomimitsu H, Ohta A, Sumida T.
Mod Rheumatol. 2019 Jan;29(1):1-19.
de Souza FHC, de Araújo DB, Vilela VS, Simões RS, Bernardo WM, Frank TA, da Cunha BM, Shinjo SK.
Adv Rheumatol. 2019 Oct 10;59(1):42.
Sunderkötter C, Nast A, Worm M, Dengler R, Dörner T, Ganter H, Hohlfeld R, Melms A, Melzer N, Rösler K, Schmidt J, Sinnreich M, Walter MC, Wanschitz J, Wiendl H.
J Dtsch Dermatol Ges. 2016 Mar;14(3):321-38.
Zanframundo et al. Arthritis Rheumatol. 2024; 76 (suppl 9).
Lilleker JB, Naddaf E, Saris CGJ, Schmidt J, de Visser M, Weihl CC; 272nd ENMC workshop participants.
Neuromuscul Disord. 2024 Apr;37:36-51. doi: 10.1016/j.nmd.2024.03.001.
Stenzel W, Mammen AL, Gallay L, Holzer MT, Kleefeld F, Benveniste O, Allenbach Y; ENMC Antisynthetase Syndrome Study Group.
Neuromuscul Disord. 2024 Dec;45:104453.
de Visser M, Carlier P, Vencovský J, Kubínová K, Preusse C; ENMC Muscle Imaging in Idiopathic Inflammatory Myopathies workshop study group.
Neuromuscul Disord. 2023 Oct;33(10):800-816.
Damoiseaux J, Mammen AL, Piette Y, Benveniste O, Allenbach Y; ENMC 256th Workshop Study Group.
Neuromuscul Disord. 2022 Jul;32(7):594-608
Mammen AL, Allenbach Y, Stenzel W, Benveniste O; ENMC 239th Workshop Study Group.
Neuromuscul Disord. 2020 Jan;30(1):70-92
Allenbach Y, Mammen AL, Benveniste O, Stenzel W; Immune-Mediated Necrotizing Myopathies Working Group.
Aggarwal R, Rider LG, Ruperto N, Bayat N, Erman B, Feldman BM, et al.
Ann Rheum Dis. 2017 May;76(5):792-801.
Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al.
Ann Rheum Dis. 2017 Feb;76(2):329-340.
19. Idiopathic inflammatory myopathies.
Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, Mammen AL, Miller FW.
Nat Rev Dis Primers. 2021 Dec 2;7(1):86. doi: 10.1038/s41572-021-00321-x.
20. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups
Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al.
Arthritis Rheumatol. 2017 Dec;69(12):2271-2282.
Lundberg IE, Fujimoto M, Vencovsky J, Aggarwal R, Holmqvist M, Christopher-Stine L, Mammen AL, Miller FW.
Nat Rev Dis Primers. 2021 Dec 2;7(1):86. doi: 10.1038/s41572-021-00321-x.
Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al.
Arthritis Rheumatol. 2017 Dec;69(12):2271-2282.
21. The wound/burn guidelines – 4: Guidelines for the management of skin ulcers associated with connective tissue disease/vasculitis
Fujimoto M, Asano Y, Ishii T, Ogawa F, Kawakami T, Kodera M, et al.
J Dermatol. 2016 Jul;43(7):729-57.
Fujimoto M, Asano Y, Ishii T, Ogawa F, Kawakami T, Kodera M, et al.
J Dermatol. 2016 Jul;43(7):729-57.
22. British Society for Rheumatology Standards, Audit and Guidelines Working Group. British Society for Rheumatology guideline on management of paediatric, adolescent and adult patients with idiopathic inflammatory myopathy
Oldroyd AGS, Lilleker JB, Amin T, Aragon O, Bechman K, Cuthbert V, Galloway J, Gordon P, Gregory WJ, Gunawardena H, Hanna MG, Isenberg D, Jackman J, Kiely PDW, Livermore P, Machado PM, Maillard S, McHugh N, Murphy R, Pilkington C, Prabu A, Rushe P, Spinty S, Swan J, Tahir H, Tansley SL, Truepenny P, Truepenny Y, Warrier K, Yates M, Papadopoulou C, Martin N, McCann L, Chinoy H.
Rheumatology (Oxford). 2022 May 5;61(5):1760-1768.
23. 239th ENMC International Workshop: Classification of dermatomyositis, Amsterdam, the Netherlands, 14-16 December 2018
Mammen AL, Allenbach Y, Stenzel W, Benveniste O; ENMC 239th Workshop Study Group.
Neuromuscul Disord. 2020 Jan;30(1):70-92.
24. French expert opinion for the management of juvenile dermatomyositis
Bader-Meunier B, Gitiaux C, Belot A, Brochard K, Mouy R, Ponce D, Bughin V, Jouen F, Musset L, Allenbach Y, Hachulla E, Maillard H, Meyer A, Bourrat E, Benveniste O; French Network of rare autoimmune, autoinflammatory diseases FAI2R.
Arch Pediatr. 2019 Feb;26(2):120-125.
25. Guidelines of the Brazilian Society of Rheumatology for the treatment of systemic autoimmune myopathies
de Souza FHC, de Araújo DB, Vilela VS, Bezerra MC, Simões RS, Bernardo WM, Miossi R, da Cunha BM, Shinjo SK.
Adv Rheumatol. 2019 Jan 22;59(1):6.
26. 224th ENMC International Workshop: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14-16 October 2016
Allenbach Y, Mammen AL, Benveniste O, Stenzel W; Immune-Mediated Necrotizing Myopathies Working Group.
Neuromuscul Disord. 2018 Jan;28(1):87-99.
Oldroyd AGS, Lilleker JB, Amin T, Aragon O, Bechman K, Cuthbert V, Galloway J, Gordon P, Gregory WJ, Gunawardena H, Hanna MG, Isenberg D, Jackman J, Kiely PDW, Livermore P, Machado PM, Maillard S, McHugh N, Murphy R, Pilkington C, Prabu A, Rushe P, Spinty S, Swan J, Tahir H, Tansley SL, Truepenny P, Truepenny Y, Warrier K, Yates M, Papadopoulou C, Martin N, McCann L, Chinoy H.
Rheumatology (Oxford). 2022 May 5;61(5):1760-1768.
Mammen AL, Allenbach Y, Stenzel W, Benveniste O; ENMC 239th Workshop Study Group.
Neuromuscul Disord. 2020 Jan;30(1):70-92.
Bader-Meunier B, Gitiaux C, Belot A, Brochard K, Mouy R, Ponce D, Bughin V, Jouen F, Musset L, Allenbach Y, Hachulla E, Maillard H, Meyer A, Bourrat E, Benveniste O; French Network of rare autoimmune, autoinflammatory diseases FAI2R.
Arch Pediatr. 2019 Feb;26(2):120-125.
de Souza FHC, de Araújo DB, Vilela VS, Bezerra MC, Simões RS, Bernardo WM, Miossi R, da Cunha BM, Shinjo SK.
Adv Rheumatol. 2019 Jan 22;59(1):6.
Allenbach Y, Mammen AL, Benveniste O, Stenzel W; Immune-Mediated Necrotizing Myopathies Working Group.
Neuromuscul Disord. 2018 Jan;28(1):87-99.
IIMS centres in ERN ReCONNET
- University Hospitals Saint-Luc
- University Hospital Ghent (adult and paediatric)
- UZ Leuven (adult and paediatric)
Helsinki University Hospital, Hospital District of Helsinki and Uusimaa, Finland (ReCONNETFIN) (adult and paedriatric)
- Civil Hospital – Brescia
- AOU Careggi, Florence
- AOU Meyer di Firenze (paediatric)
- IRCCS AOU San Martino – Genoa
- Foundation IRCCS CA’Granda Ospedale Maggiore polyclinic – Milan
- IRCCS Ospedale San Raffaele di Milano
- ASST Centro Specialistico Ortopedico Traumatologico Gaetano Pini (Presidio Pini)
- AO Padua
- Foundation IRCCS Polyclinic San Matteo, Pavia
- AOU Pisan
- AOU Policlinico Umberto I di Roma
- Fondazione Policlinico Tor Vergata Roma
- A.S.L. Torino – Hub S. Giovanni Bosco
- Azienda Ospedaliera Universitaria Integrata di Verona
- University Medical Centre Ljubljana (paediatric)
- Hospital Clínic de Barcelona y Hospital de Sant Joan de Déu (adult and paediatric)
- Hospital Universitari Vall d’Hebron (adult and paediatric)
- Hospital Universitario 12 de Octubre
Karolinska Universitetssjukhuset (adult and paediatric)
